Research
Our research can be divided into five areas (click on a theme to scroll down):- Ultrafast dynamics of chromophores and the impact of solvation
- Isomer-specific spectroscopy and photoisomerisation dynamics
- 2D photoelectron-photodetachment spectroscopy of temporary anion and non-valence states
- PAHs and carbonaceous molecules in space; radiative stabilisation mechanisms
- Quantum chemical calculations and ab initio molecular dynamics to support experiments
We build and operate custom, ultrahigh vacuum mass spectrometry and ion spectroscopy and tunable laser systems to study isolated molecules with high temporal and spatial resolution. Our instruments (two main instruments, and one under slow construction) are descibed on the Instruments page. We are advocates of Autodesk Inventor and 3D printing to lower the cost and simplift instrument development.
Ultrafast dynamics of chromophores and the impact of solvation

Chromophores are light-absorbing molecular units, encompassing aromatic groups, dyes, and other highly conjugated systems. In biological contexts, biochromophores serve as the photoactive unit in biomolecules, including the chromophores in fluorescent proteins (e.g., green fluorescent protein), photoactive yellow protein, flavins, retinoids, and photoprotective pigments. These biochromophores usually have structural diversity, existing in multiple isomeric, tautomeric, and protonation states, each with distinct photophysical and photochemical behaviors. We wish to separate these and investigate their individual role. Understanding the interplay between these forms is key to elucidating the mechanisms underlying biological light absorption and energy conversion.
Gas-phase studies provide a uniquely controlled environment for investigating the intrinsic photophysics of biochromophores, free from the perturbations introduced by solvation or the surrounding protein matrix. By isolating the chromophore, one can directly probe fundamental excited-state processes such as internal conversion and photoisomerisation on their native potential energy surfaces. The solvent-free conditions also enable rigorous comparisons with ab initio electronic structure and non-adiabatic dynamics calculations, as the absence of environmental effects simplifies the theoretical description. Together, high-resolution gas-phase spectroscopy and state-of-the-art computational methods yield a detailed molecular-level understanding of how structure governs light-induced dynamics in biologically relevant chromophores.
Isomer-specific spectroscopy and photoisomerisation dynamics
2D photoelectron-photodetachment spectroscopy of temporary anion and non-valence states
Velocity-map imaging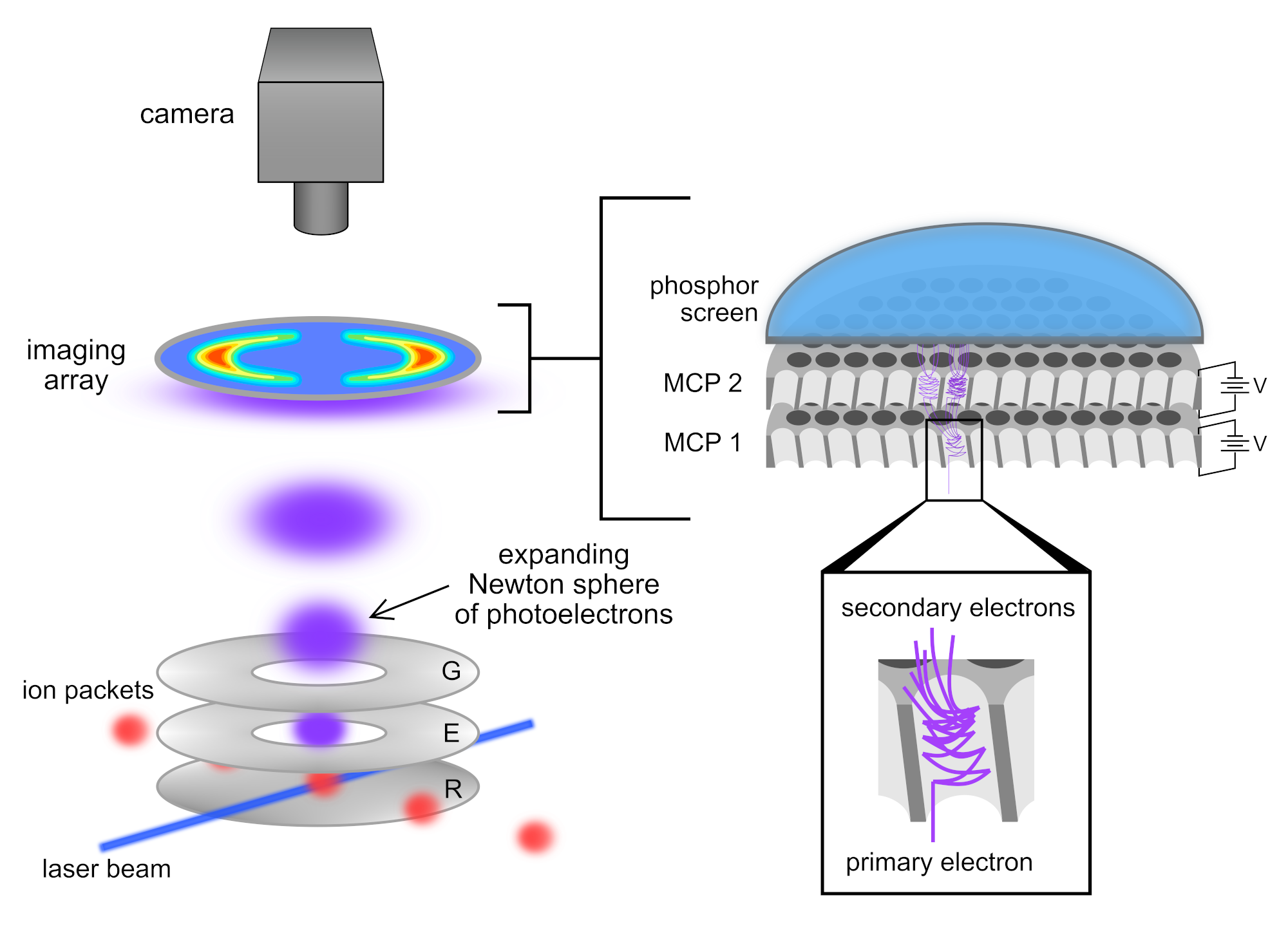
PAHs and carbonaceous molecules in space; radiative stabilisation mechanisms
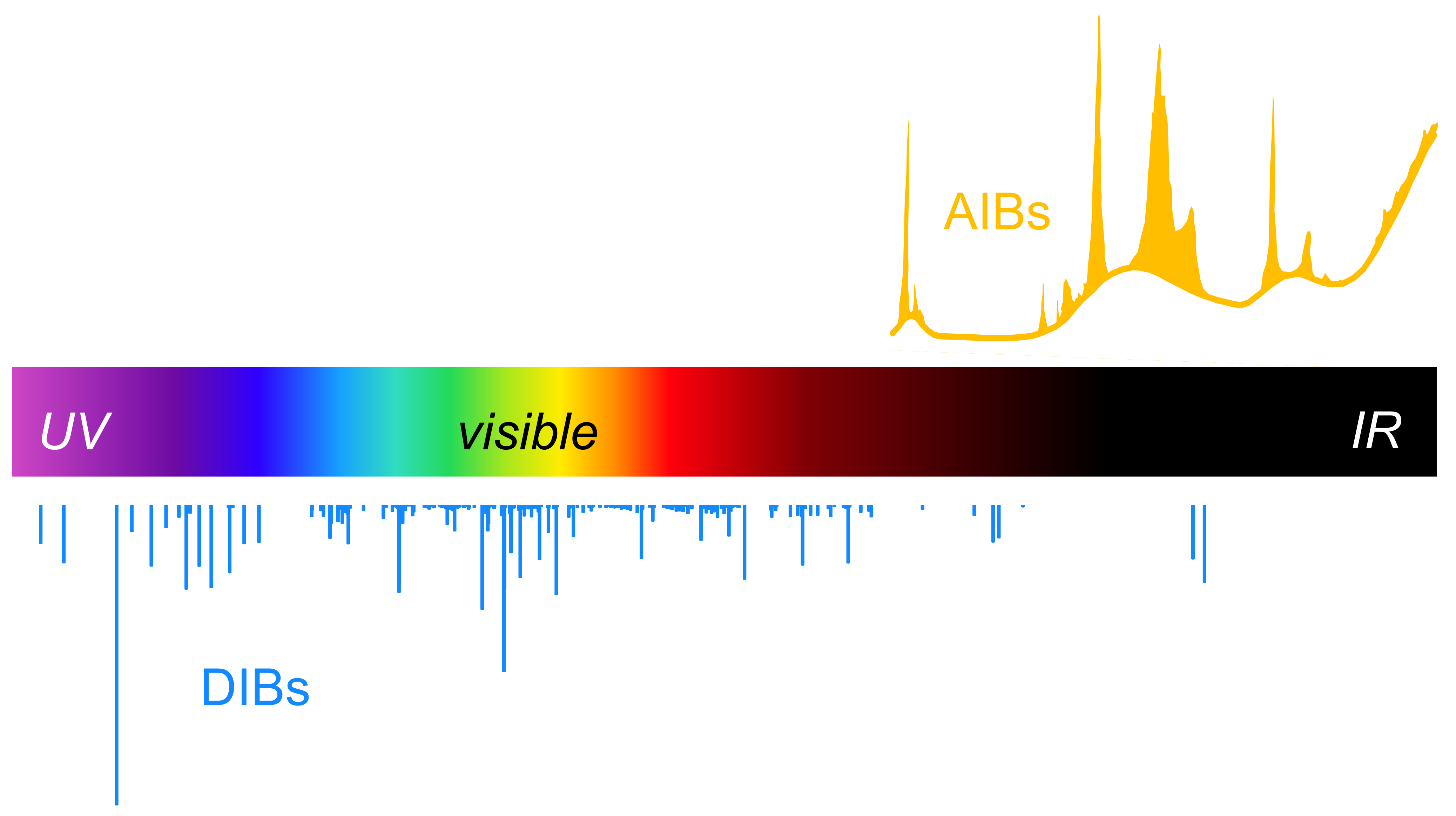
Polycyclic aromatic hydrocarbons (PAHs) are thought to be one of the major reservoirs of carbon in the interstellar medium. This hypothesis is supported by widespread astronomical observations of unidentified infrared bands (UIBs), now largely interpreted as aromatic infrared bands (AIBs) due to their strong resemblance to the infrared (IR) spectra of aromatic and PAH molecules. The recent deployment of the James Webb Space Telescope, with its unprecedented sensitivity in the infrared region, is further revealing the ubiquity of these IR emitters across a wide range of astrophysical environments. In collaboration with Dr. Mark Stockett at Stockholm University, our research focuses on elucidating the underlying physics and chemistry of PAHs and other carbonaceous molecules that have been detected through radioastronomy, as well as exploring potential candidates for the many still-unidentified molecular features observed in space through their potential stability mechanisms.
This research theme utilises a range of high-vacuum, gas-phase experimental platforms at specialised facilities, including photoelectron–photoion coincidence (PEPICO) spectroscopy at synchrotron facilities, the DESIREE infrastructure at Stockholm University, which is an electrostatic ion storage ring capable of storing and cooling ions at cryogenic temperatures, and other infrared spectroscopy techniques. These techniques allow us to probe the intrinsic photophysics and stability of PAHs, typically in their ionic forms, under controlled conditions. In particular, our recent work using DESIREE has focused on understanding the interplay between infrared emission and recurrent fluorescence as competing radiative relaxation pathways that contribute to the long-term stability of PAHs following excitation through collisions or absorption of visible or near-UV photons. Recent molecules of interest include the indenyl cation, 2-cyanoindene, and 1-cyanonaphthalene.
Quantum chemical calculations and ab initio molecular dynamics to support experiments
Gas-phase experiments provide critical reference data for calibrating and benchmarking quantum chemical and dynamical simulations of photoisomerising systems. By isolating molecules (ideally as anions) from environmental perturbations, these measurements yield well-defined observables, including electron detachment, excitation energies, and isomerisation dynamics (yields, branching ratios), that can be directly compared with ab initio calculations. Such comparisons are essential for assessing the accuracy of theoretical models and guiding the development and refiement of improved methods.
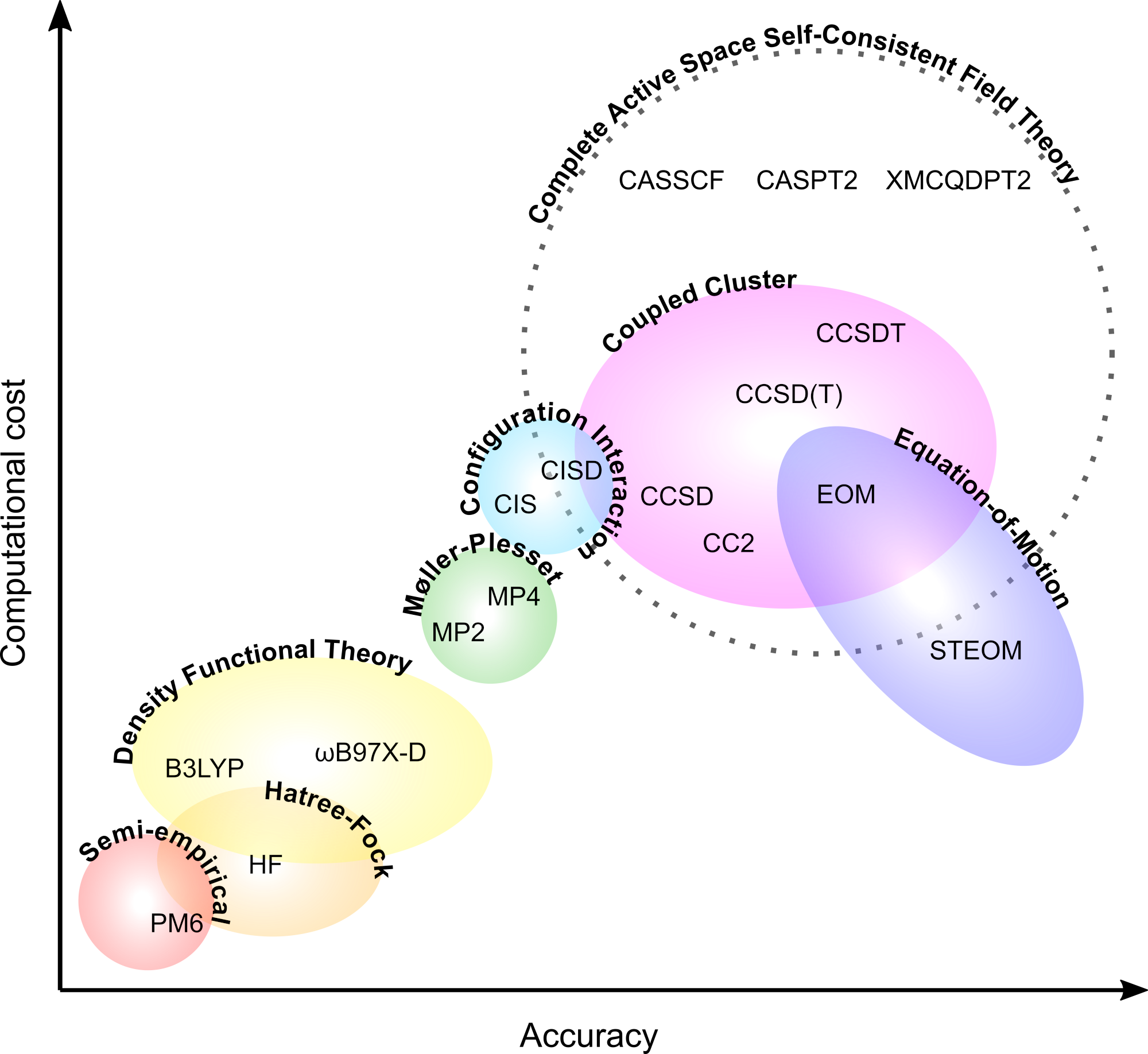
We employ a range of electronic structure packages, including ORCA, GAMESS-US, and OpenQP, to model the ground- and excited-state properties of photoswitches, biochromophores, polycyclic aromatic hydrocarbons (PAHs), and other molecular systems of astrochemical and biological relevance. We are actively pursuing the use of multireference spin-flip time-dependent density functional theory (MRSF-TDDFT) to describe photoinduced isomerisation processes in photoswitches and biochromophores, alongside surface-hopping nonadiabatic molecular dynamics simulations that allow for direct comparison with our experimental observations.
Our theoretical work is driven by experimental results. The ability of our experimental techniques to monitor isomerisation dynamics with high structural sensitivity places us in a unique position to test and refine theoretical descriptions of nonadiabatic transitions and relaxation pathways. This synergy between experiment and computation enables a comprehensive understanding of light-induced molecular dynamics from first principles.